(1)The side chain protectors of the bridged two sites cysteine must be the same to ensure simultaneous removal while oxidizing to disulfide bonds;
(2)When two pairs of disulfide bonds are contained, except that the protective bases of each pair of bridgehead cysteines are the same, the side chain protective groups of cysteines that are not linked to each other should be different, and their removal conditions should be in a normal way that does not disturb each other.
(3)The side chain protector removal conditions of all cysteines should not allow Linker to undergo a cleavage reaction.
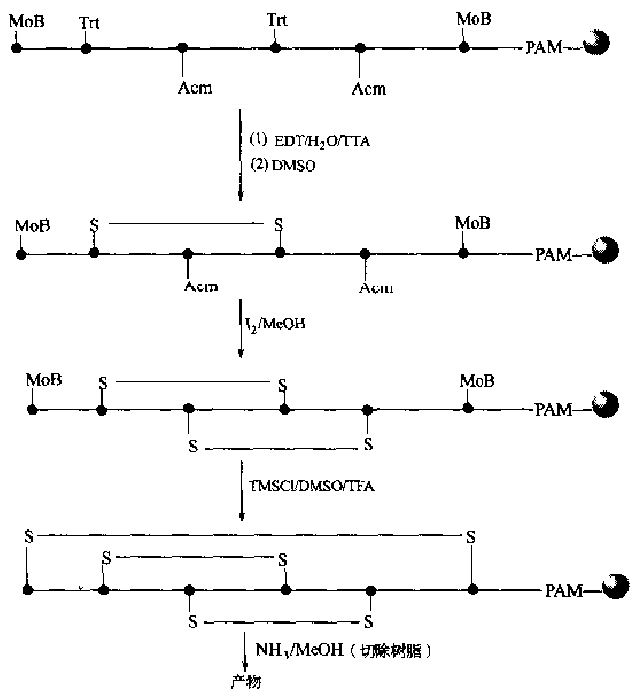
Due to the characteristics of solid-phase synthesis, besides the requirement for side chain protecting groups of Cys, attention should also be paid to oxidants, which are soluble in organic solvents, such as I2, DMSO and other conditions.Potassium ferricyanide [K3FeCN)6], glutathione system GSH/GSSG, etc., which are commonly used in liquid phase reactions, cannot be used for solid phase oxidation to generate disulfide bonds because of their insolubility in organic solvents.It is difficult to synthesize two or more disulfide bonds on the same peptide chain and ensure correct pairing.An example is given to illustrate how this synthesis is accomplished in a solid phase manner (Fig.
Reaction route for solid-phase generation of 3 pairs of disulfide bonds</center>
2、vLactam-type cyclic peptide
Compared with the synthesis of disulfide-bonded cyclic peptides, the side chain protecting groups of amino and carboxyl groups in the synthesis of phthalamine-bonded cyclic peptides are much simpler than those in the case of Cys.Another feature is that the position of bridging is mostly in the way that N-terminal residues of the peptide chain are connected with C-terminal residues, i.e., head-to-tail.In the synthesis of liquid-phase head-to-tail cyclic peptides, as in conventional condensation-conjugation peptide reactions, the carboxyl activation conditions, i.e., the involvement of condensation agents, are required.Because the C-terminal residues in solid-phase peptide synthesis are bonded to the carrier Linker, they cannot be activated with a mixture during cyclization.In this case, ester-bonded Linker: Aminolysis by N-terminal-NH2 should be the basic way to prepare amide-type cyclic peptides head-to-tail in solid phase.Although theoretically all ester-bonded Linkers can undergo intramolecular aminolysis.However, except for the generation of cyclic dipeptides in the form of six-membered ring DKP, only oxime-type Linker resin can easily react with the N-terminal amino group of the peptide chain to generate cyclic peptides:
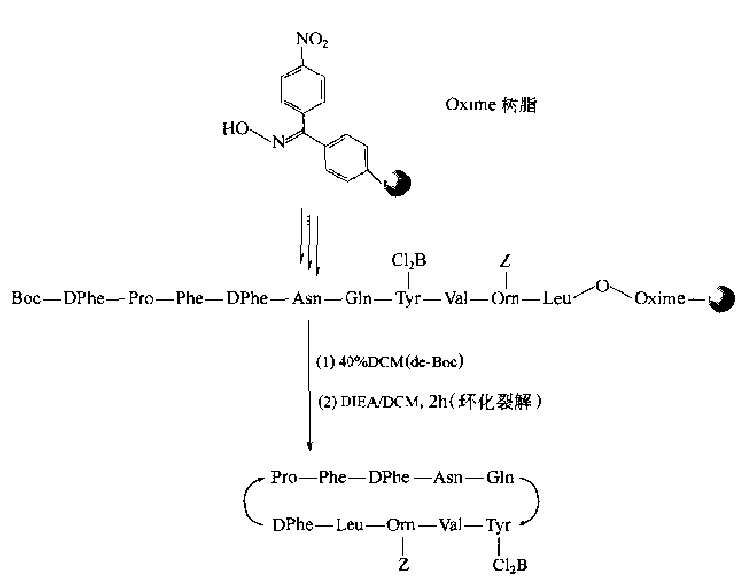
Obviously, the activity of other ester linker bonds is not sufficient for intramolecular aminolysis beyond the six-membered ring.If there is another free -COOH group on the peptide chain (specially designed in the synthesis).The amide-bonded cyclization can occur under the action of the condensant.To this end, the structural characteristics of Glu, Asp and other residues in the target stomach must be fully utilized in the design and synthesis, i.e. to try to bond their side chain active groups to the Linker of the carrier.The remaining alpha-amino and alpha-carboxyl groups can extend the peptide bi-directionally or unidirectionally.When the target sequence is assembled, the N and C ends can be bonded to synthesize the ring with the activation of the condensant (Fig.
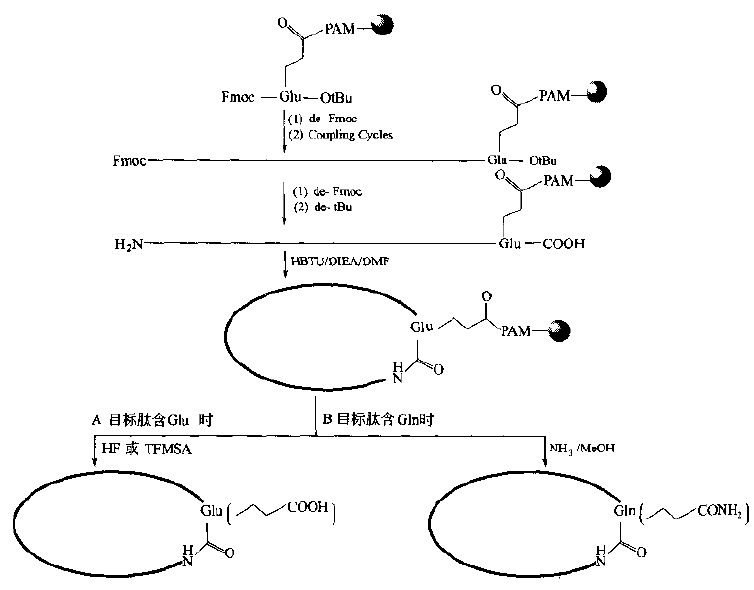
Synthesis of Cyclic Peptides Linked to Carriers by Side Chain-C00H
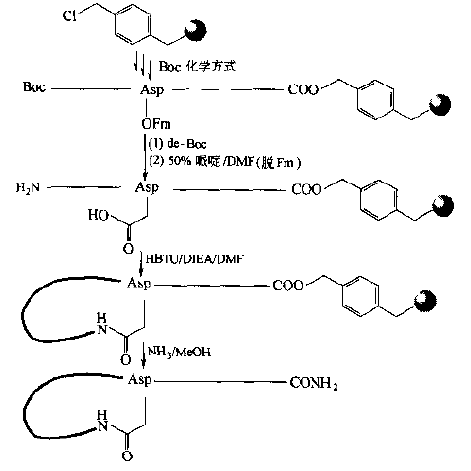
In addition to Fmoc chemistry, Boc chemistry can also be used, but the ASP or Glu of the ring-forming sites still need orthogonal protection strategies.For example, the side-chain free carboxyl groups of Boc-AsP-OFm or Boc-Glu-OFm are linked to the carrier, the Boc is removed first, then the peptide bond is extended on the amino side, and finally the OFm is removed, then the free COOH is activated, and molecular lactam bond cyclization occurs.This way the Asp or Glu residues can be located in the middle of the peptide chain.That is, not linked to the carrier, resulting in an amide-bonded cyclic peptide in the form of a "head-to-head" (Fig.
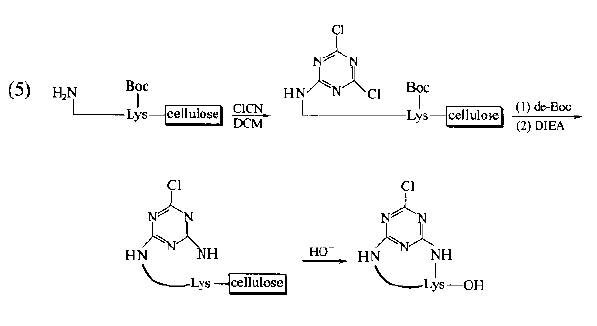
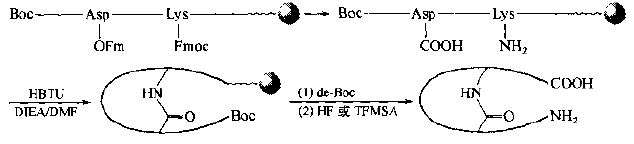
Another way to synthesize "mid-linking" cyclized peptides is to introduce one Lys-1 (Fmoc) and one Asp (OFm) or one Glu (OFm) into the peptide chain as two bridge residues:
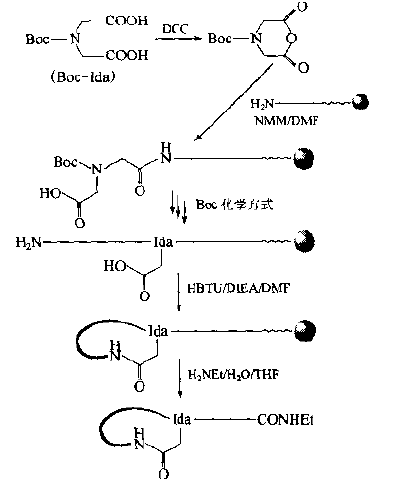
There is also a method in which iminodiacetic acid (Ida) is used as a member to provide a bridge head-COOH group to form an amide-bonded cyclic peptide in the form of a "head-in" with the N-terminal-NH3 group.This synthesis is characterized by the absence of amino acids protected in any orthogonal manner without additional Fmoc or OFm removal operations.
二、Nonclassical cyclic peptide:
1、depsipeptide
Generally speaking, the synthesis method suitable for amide bond can form molecular lactone bond.As with the amide cyclopeptides described earlier, the selection of bridgehead residues and their side chain protection is the key.As one of the precursors of acyl bond, the carboxyl components are ASP (otbu) I, - Glu (otbu) I, ASP (OFM) I and OFM I. The first two are suitable for Fmoc protection strategy and the second for BOC protection strategy.Another precursor hydroxy component of an ester may use a non amino acid component, such as a hydroxy acid or a phenolic hydroxy carboxylic acid, but they are only suitable for positioning at the N-terminal:
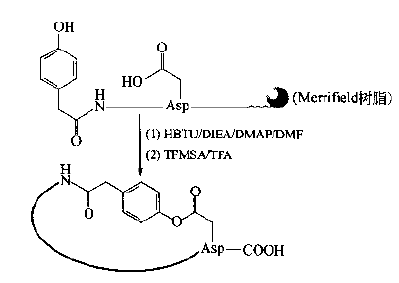
The more commonly used hydroxyl component is the use of any of the three natural amino acids ser, thr and Tyr.However, their side chain protecting groups should be removed in an orthogonal way with the α - amino protecting groups at the N-terminal, so as to avoid the interference of α - amino group on the esterification cyclization.In addition, they can be placed anywhere in the skin chain:
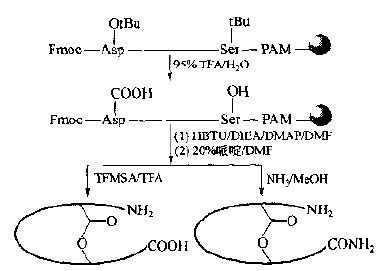
It should be noted that the practical scope of this cycloester synthesis is relatively limited, because there should be no Lys, Ser, Thr, Tyr and Asp in other locations except the bridgehead.This is because their side chain protectors do not have enough other orthogonal protection and removal methods (while Cys has many orthogonal protectors), which interferes with the bridging ring of bridgehead residues, so the structural design of the target peptide will be more limited.
2、Monosulfide cyclic peptide
It was found that the metabolism of single thioether cyclopeptide was slower than that of disulfide cyclopeptide, and most of them were non immunogenic.The solid-phase synthesis methods of these cyclopeptides are as follows:
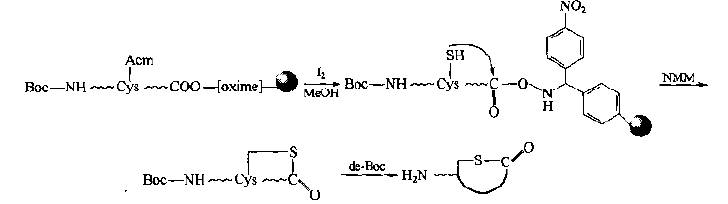
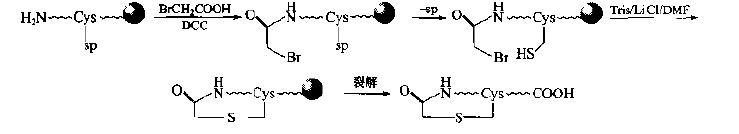
(1) PCORR method(peptide cyclization on oxime resin) In this way, the linker nucleophile of oxime is very sensitive to attack, and the peptide chain is introduced into Cys. After the protection is removed, the free SH group attacks linker bond, forming a single thioether ring skin. At the same time, linker cracking occurs to remove the carrier.In this case, the N-terminal α - amino group should be protected, otherwise, the linker will also undergo amination reaction.Therefore, the conditions of removing Cys side chain protecting group and a-amino group should be orthogonal:
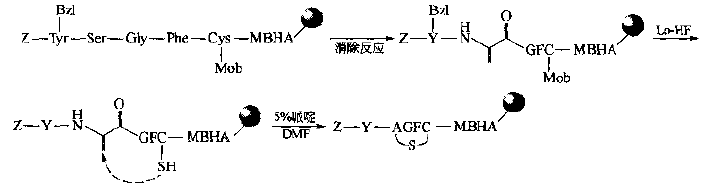
(2)Michael addition method is used to eliminate the ser residue at the appropriate site in the peptide chain, and generate DEHYDROALANINE (DHA). The latter is added with the free SH group on the peptide chain by Michael to obtain the single thioether cyclopeptide. Finally, the side chain protection is removed and the carrier is removed.This way is also considered as a way to simulate biosynthesis.
Another way to prepare the monophthioether cyclopeptide through Michael addition is to introduce acrylic acid at the N-terminal of the peptide chain, and then carry out Michael addition to obtain the "head to middle" or "head to tail" type cyclopeptide:
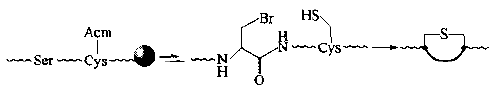
(3)In addition to dehydrating to form DHA for Michael addition, the ser in the nucleophilic substitution peptide chain can also convert its side chain - oh to a br, and then undergo intramolecular nucleophilic substitution reaction with an SH group side chain on the peptide chain. In the same way, a single thioether cyclic peptide with ALA and Cys as two bridge residues is obtained:
If the expected product is a "head to middle" or "head to tail" type of single thioether cyclic peptide, it can introduce human bromoacetyl structure at the N-terminal, and then carry out intramolecular nucleophilic substitution.This method is more simple and does not need the structural transformation of residues:
3.Oxygen and nitrogen bridged cyclopeptides
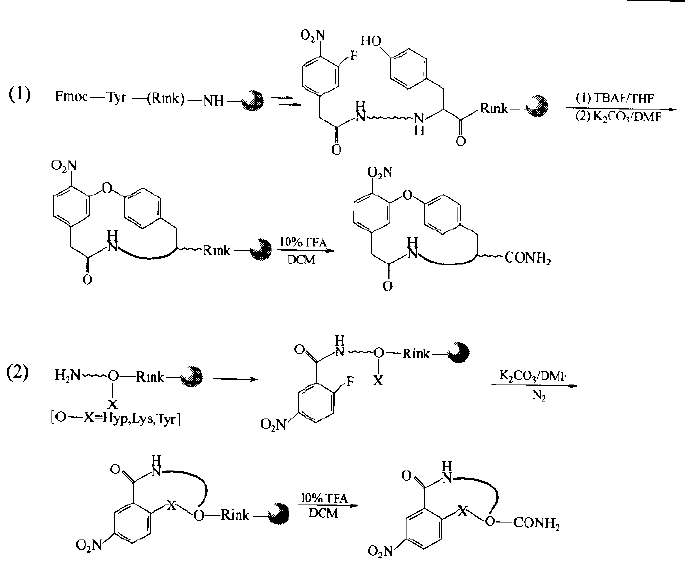
N-terminal human halophthalein can be substituted with side chain groups of Tyr, Lys, Arg, his and other residues in the peptide chain by intramolecular nucleophilic substitution.For example, an O-type cyclopeptide is generated by reacting with the phenol hydroxyl of Tyr, and an nh-type cyclopeptide is generated by reacting with the side chain amino of Lys.It should be noted that the same chain should not contain more than two sites with active side chain bases.
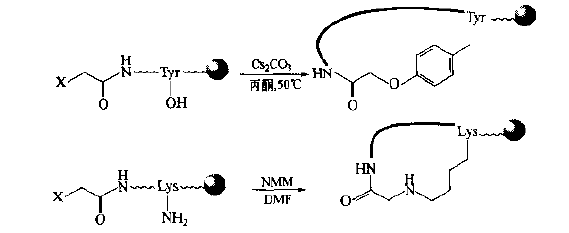
4.Mannich Alkaline cyclic peptide
If there is Tyr residue in the peptide chain, the α position of the phenolic hydroxyl can be used as the donor of active hydrogen to form Mannich condensation with amines and aldehydes.In fact, the N-terminal amino group of peptide chain is the ready-made amine component.Mannich basic cyclopeptide can be prepared by adding aldehyde component.
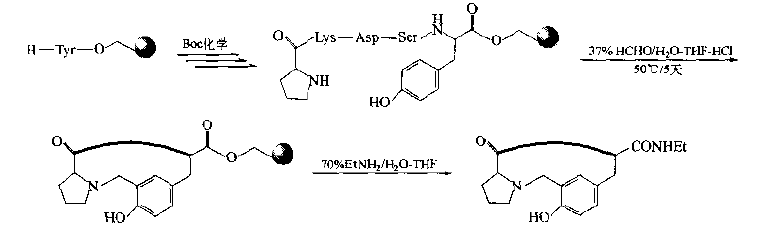
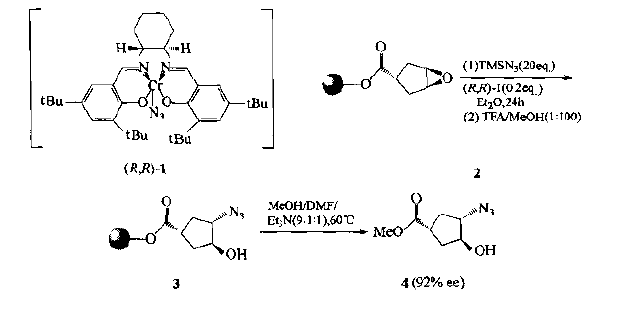
5.Template intervening cyclopeptide
It was found that the racemic epoxide (2) bonded on the solid support could asymmetric catalyze the ring opening under the action of trimethylazide silane (tm-sn3) and chromium-n3 complex (1), resulting in the stereoselective product trans azide Cyclopentanol (3):
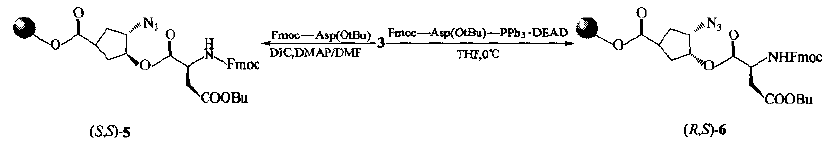
RGD} peptide was synthesized by using chiral template 3 as scaffold.There are two kinds of condensation ways when aspartic acid and 3-hydroxy-ester are connected on the same day: 1) conventional condensation agent DIC is used to obtain 5 with constant configuration, i.e. it is still trans disubstituted; 2) aspartic acid formed by Mitsunobu reaction condition is cis-6 with azido.
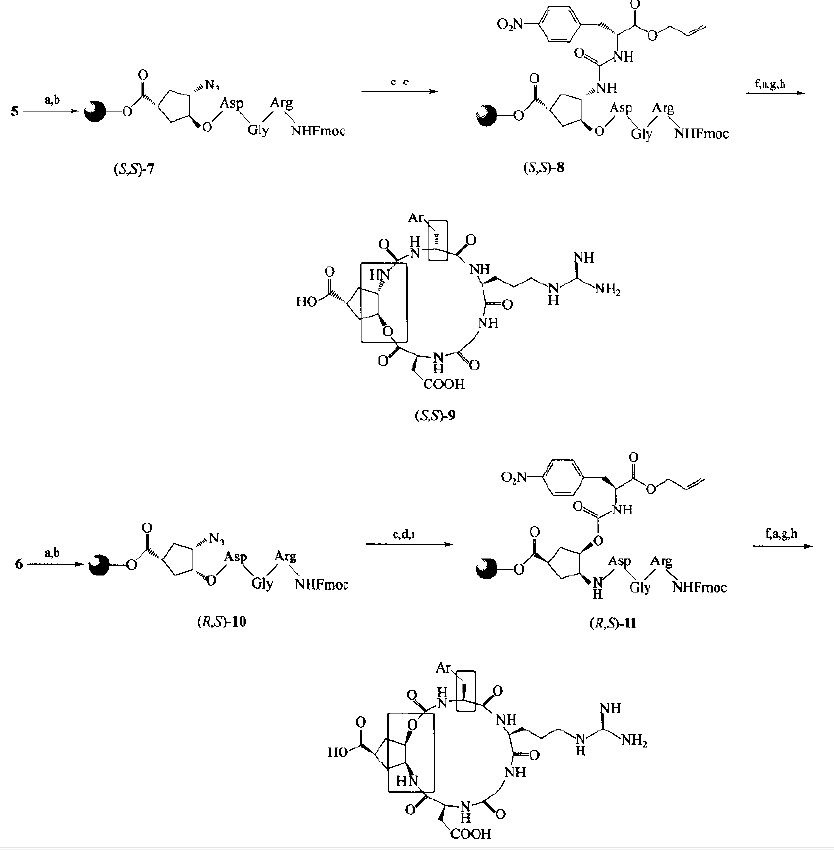
Taking 5 and 6 as the starting materials, after the peptide chain assembly, the azido group was converted into carboxyl precursor structure 8 and 11, respectively, and finally the cyclic peptide was synthesized with α - amino condensation of arg:
6,Aromatic cyclopeptide
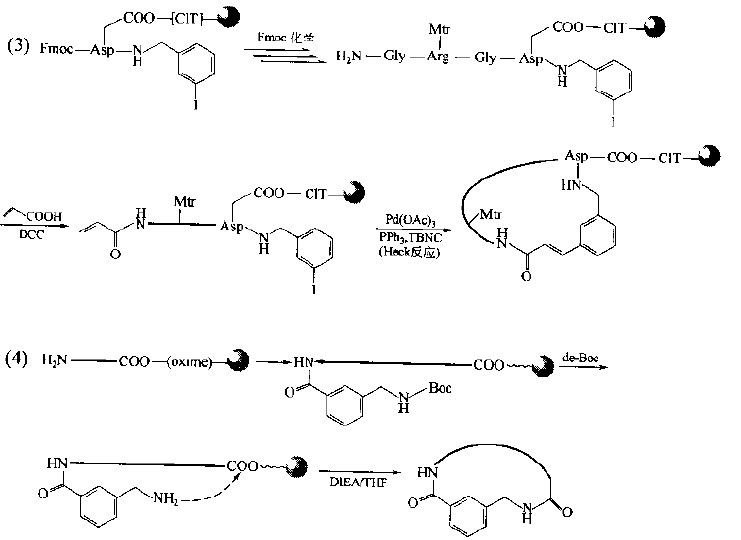
Cyclic peptides with general bridging structure have improved the conformational flexibility of straight chain peptides.It has been found that if the bridging structure mediates the aromatic ring structure, the conformational qualification of this kind of cyclopeptide will be further enhanced due to the flatness requirements of the latter, so the selectivity of its binding to the receptor will be improved and the biological activity will be enhanced.The synthesis of this kind of ring requires an aromatic ring derivative containing double active functional groups as a bridge component, and each functional group is bonded to the bridgehead residue on the peptide chain respectively.In the following synthetic examples, fluorobenzoic acid, iodobenzylamine, aminomethylbenzoic acid and cyanuric chloride are used as the bridged precursors.